

Scientists at the Vilnius University Life Sciences Center (VU LSC) investigate the structures of biological molecules, their interactions, and develop methods to assess the reliability of computationally predicted structures.
In recent years, artificial intelligence–based tools have fundamentally transformed the ability to predict protein structures. The most prominent example is the AlphaFold model, whose developers were awarded the Nobel Prize in Chemistry in 2024. Nevertheless, even the most advanced methods do not always enable accurate prediction of all biomolecular structures, leaving scientists still to evaluate computational models reliably.
Structural biology today: what scientists study and why
Research into the structures of biological molecules allows scientists to understand how life functions. It is the shape of molecules and their interactions that determine many processes within the cell.
“Virtually all processes in the cell occur because different molecules interact. By binding together, they form various biomolecular complexes – from small assemblies of just a few molecules to very large structures such as ribosomes, which are responsible for protein synthesis,” explains Dr. Justas Dapkūnas, a scientist at the Vilnius University Life Sciences Center.
According to him, understanding these structures is important for two main reasons. First, it helps reveal the fundamental principles of how life works. Second, structural information is essential for developing new drugs and products in the chemical industry.
“Drug molecules often act by binding to specific natural biological molecules. Both small organic compounds and larger proteins, such as antibodies, are used as drugs. Therefore, knowing their structures is critically important.
In some cases, enzymes are used as catalysts in the chemical industry. Still, natural proteins are not optimised for industrial conditions, as they have evolved to function only within living organisms. Understanding their structures allows us to modify them,” the scientist says.
How were protein structures studied before artificial intelligence?
Determining protein structures experimentally is often complex, expensive, and time-consuming. For this reason, scientists have long sought to predict these structures using computational methods.
“For a long time, researchers attempted to predict protein structures based on the principles of physics and chemistry, but this was not very successful. The fundamental principles governing protein folding are still not fully understood,” says Dr. Dapkūnas.
Better results were achieved by applying biological principles. “It was observed that evolutionarily related proteins, even when found in different organisms, often share similar structures. For a long time, one of the most reliable methods for predicting protein structure was homology modelling – inferring a structure based on a known structure of a similar protein.
Another important principle is co-evolution. If certain amino acids change together in related proteins, they are likely close to each other in the three-dimensional structure,” he explains.
The analysis of such coordinated changes allows researchers to determine which amino acids are spatially close within a protein structure. This became one of the key early approaches to structure prediction.
How is the accuracy of structure prediction assessed?
For several decades, scientists have tested their methods in international experiments – CASP (Critical Assessment of Structure Prediction) and CAPRI (Critical Assessment of Predicted Interactions). These initiatives evaluate protein structure prediction and biomolecular interaction modelling.
“In these experiments, scientists are given structures that have already been determined experimentally but have not yet been made public. This allows for an objective assessment of how accurately different methods can predict structures,” says Dr. Dapkūnas.
The Bioinformatics Department at the Vilnius University Life Sciences Center regularly participates in these competitions and, in recent years, has been among the most successful.
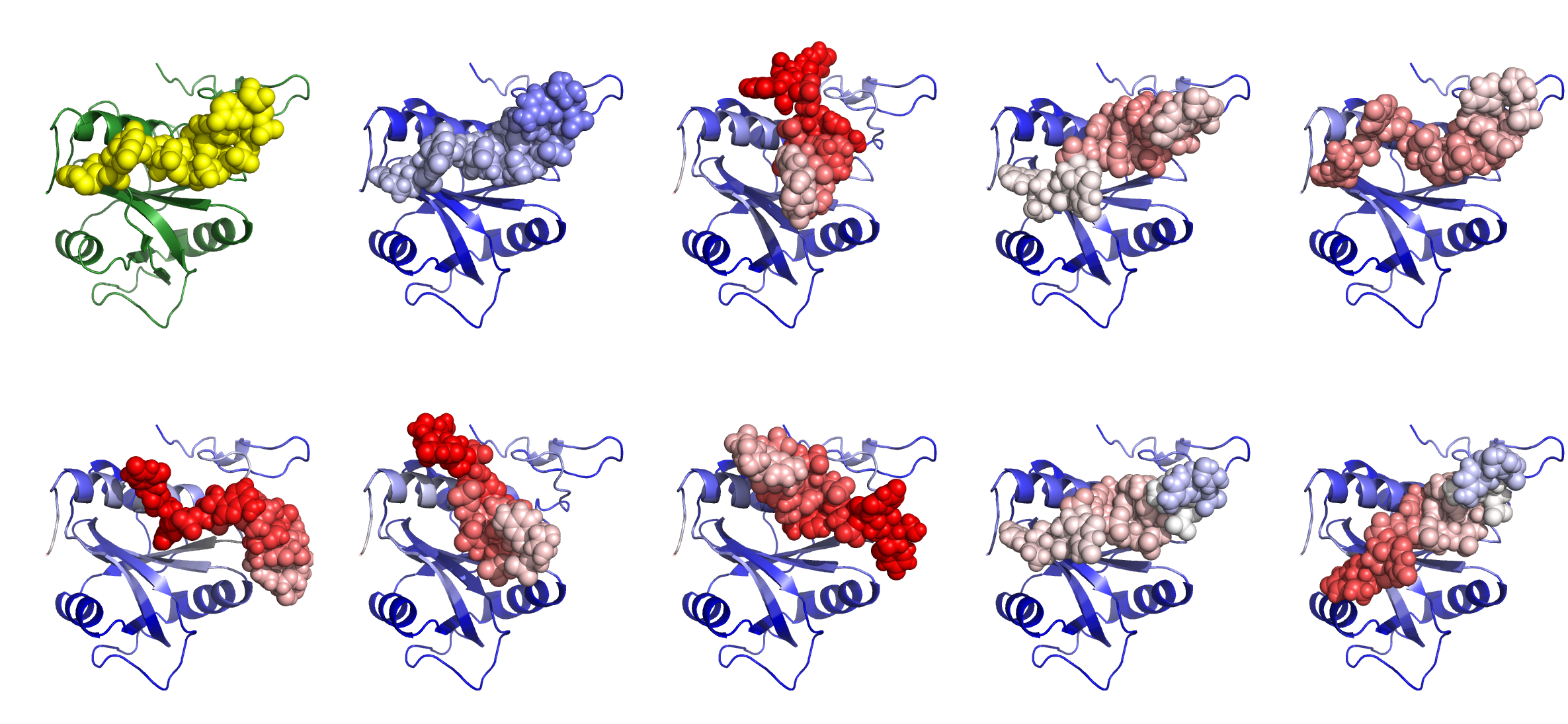 |
|
The experimentally determined structure of the protein–RNA complex (PDB: 6FPQ) is shown in green and yellow. |
The AlphaFold breakthrough
AlphaFold has driven the greatest revolution in structural biology in recent years. “AlphaFold 2 was the first method capable of predicting protein structures with a level of accuracy that, in many cases, rivals experimental approaches,” says Dr. Justas Dapkūnas.
The method was introduced in 2020 and tested in the CASP14 experiment. Its success was made possible by combining approaches from several fields – structural biology, bioinformatics, and artificial neural networks.
According to the scientist, there was no single moment when artificial intelligence entered structural biology. “Machine learning methods had been used in this field for many years, but eventually enough data accumulated and neural network techniques improved,” he explains.
Where do the limits of artificial intelligence lie?
Today, AlphaFold and similar methods can predict the structures of many proteins with high accuracy. “In many cases, scientists no longer need to model proteins themselves – their structures have already been predicted and deposited in public databases,” says Dr. Dapkūnas.
However, not all molecules can be predicted with equal accuracy. More challenging cases include protein–small molecule interactions, antigen–antibody complexes that are particularly important for the pharmaceutical industry, as well as DNA and RNA structures and large biomolecular complexes, which are more often associated with fundamental research questions.
“Artificial intelligence tends to predict structures more accurately when they are similar to those used during model training. However, entirely new types of interactions still pose difficulties,” explains doctoral researcher Rita Banciul.
In addition, neural networks operate as so-called “black boxes.” “Such models may contain millions or even billions of parameters, making it difficult to determine exactly why they arrive at a particular decision. This lack of transparency makes it harder to fully trust the results, especially in applications that require extremely high structural accuracy,” she adds.
The contribution of Vilnius University Life Sciences Center scientists
Researchers at the Vilnius University Life Sciences Center aim to address some of these challenges by developing new methods for evaluating biomolecular structures.
“One of the key challenges is that different modelling tools can generate many alternative structures. It is therefore essential to have methods that can assess their quality and help identify the most realistic model,” says R. Banciul.
To address this, a structure quality assessment tool, VoroMQA-aa, is being developed within the framework of the Research Council of Lithuania–funded project “Deep learning-based methods for annotating protein interactions” (S-MIP-23-44).
“In real biomolecules, atoms are not arranged randomly – their contacts follow the laws of physics and chemistry. Our method analyses these contacts and allows us to determine whether a predicted structure is realistic,” explains R. Banciul.
The method is based on the principle of Voronoi tessellation, which allows the space around each atom to be mathematically defined and interatomic contacts and their surface areas to be calculated.
“If a model contains atomic contacts that are commonly observed in real structures, it receives a high quality score. If the contacts are unusual or rarely observed in experimentally determined structures, the score decreases,” she says.
Why are such methods important?
According to R. Banciul, structure quality assessment methods become particularly important when modelling complex systems.
“Sometimes, hundreds or even thousands of different models are generated for a single system. Among them, there is often a very accurate one – but it needs to be identified. This is where structure quality assessment tools are essential,” she notes.
VoroMQA-aa can evaluate models of various biomolecules, including proteins, nucleic acids, and protein–ligand complexes.
“It is also important that our method is not based on deep learning. This allows it to assess models generated by artificial intelligence independently and reduces the risk of misinterpretation,” emphasises R. Banciul.