
 |
ČESLOVAS VENCLOVAS Distinguished Professor
|
|

Protein Structural Bioinformatics
Proteins typically function as three-dimensional (3D) structures, often through interaction with each other and/or with other macromolecules. Protein 3D structure is also the most conserved property of evolutionary related proteins. Therefore, the knowledge of structures of individual proteins and their complexes is essential for understanding their evolution, function and molecular mechanisms. However, the experimental determination of protein structure is slow, expensive and not always successful. The increasing computer power and the flood of biological data make computational prediction of 3D structure of proteins and their complexes an important alternative to experiments. Computational methods are also indispensable in the analysis or prediction of interaction sites even in the case of experimentally solved structures. However, computational methods have their own challenges. Computational structure prediction works best when related structures (templates) are available. Therefore, the detection of remote homology is one of the major impediments. The reliable estimation of the accuracy of predicted structures is another important problem. More efficient methods for the analysis and prediction of protein binding sites are also badly needed.
Our team addresses a broad range of protein-centred research topics that can be collectively described as Computational Studies of Protein Structure, Function and Evolution. There are two main research directions:
1) Development of computational methods for detection of protein homology, for comparative protein structure modelling, and for analysis and evaluation of 3D structure of proteins and protein complexes. In recent years, we have developed several new methods addressing these research topics. All of the software packages implementing these methods are freely available at our web site (http://bioinformatics.lt/software).
2) Application of computational methods to biological problems. In this research direction, we have been using computational methods for discovering general patterns in biological data, structural/functional characterization of proteins and their complexes, design of novel proteins and mutants with desired properties. Over the years, our major focus has been on studies of DNA replication and repair systems in viruses, bacteria and eukaryotes. In addition, we have entered a highly dynamic CRISPR-Cas research field and have already made important contributions in elucidating structural and mechanistic properties of CRISPR-Cas systems and their evolutionary relationships.
SELECTED PUBLICATIONS
1. Makarova et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020, 18(2): 67–83. doi:10.1038/s41579-019-0299-x.
2. Gasiunas et al. A catalogue of biochemically diverse CRISPR-Cas9 orthologs. Nat Commun. 2020, 11(1): 5512. doi:10.1038/s41467-020-19344-1.
3. Kazlauskas et al. Diversity and evolution of B-family DNA polymerases. Nucleic Acids Res. 2020,; 48(18): 10142–10156. doi:10.1093/nar/gkaa760.
4. Dapkūnas, J., Olechnovič, K. & Venclovas, Č. Structural modeling of protein complexes: Current capabilities and challenges. Proteins. 2019, 87(12): 1222–1232. doi:10.1002/prot.25774.
5. Olechnovič, K. & Venclovas, Č. VoroMQA web server for assessing three-dimensional structures of proteins and protein complexes. Nucleic Acids Res. 2019, 47(W1): W437–W442. doi:10.1093/nar/gkz367.
Predicting Structures of Protein Assemblies in Global CASP and CAPRI Experiments
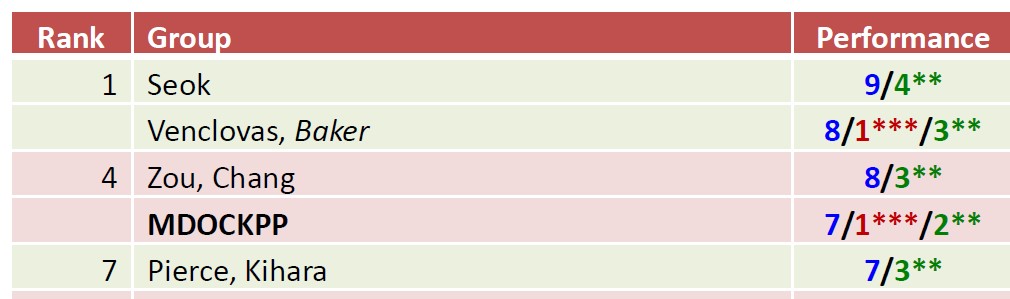
Measuring progress in ability to predict 3D structures of protein complexes is the goal of CAPRI experiments and, lately, one of the major aims of CASP experiments. In summer of 2020, we participated in both CASP14 and CAPRI experiments that were executed in parallel. In both experiments, we tested the performance of our template-based modelling, free docking and hybrid modelling protocols. Among key components of these protocols were the latest versions of PPI3D and VoroMQA methods, developed in our group. The PPI3D web server enables searching, analysing and modelling protein complexes, whereas VoroMQA allows estimation of protein structure quality. Independent assessors found our CAPRI results (group ‘Venclovas’) to be at the top jointly with the results of the other two groups. According to CASP assessment, our group was second. CASP and CAPRI experiments were performed on different test sets, and assessment methods were somewhat different. Despite slight differences in overall ranking, our group continued to be one of the leading groups in the modelling of protein complexes. The results of the joint CASP14-CAPRI experiment are to be published in a special issue of Proteins.
Uncovering Convoluted Evolutionary History of Human Replicative Polymerases
B-family DNA polymerases (PolBs) are the most common replicases, present in all domains of life and in many DNA viruses. Despite extensive research, origins and evolution of PolBs remain enigmatic. To unravel the evolutionary history of PolBs, we performed comprehensive computational analysis of these proteins originating from archaea, bacteria, eukaryotes and viruses. As a result, we defined and characterized six new groups of archaeal PolBs and a new group of bacterial PolBs, which appears to be related to the catalytically active N-terminal module of the eukaryotic Polε. We also uncovered the similarity of the catalytically inactive Polε C-terminal module to Polα. Finally, we discovered that two novel groups of archaeal PolBs have C-terminal metal-binding domains, closely related to those present in eukaryotic Polα and Polε. Collectively, the results of this study allowed us to propose a scenario for the evolution of human and other eukaryotic PolBs.
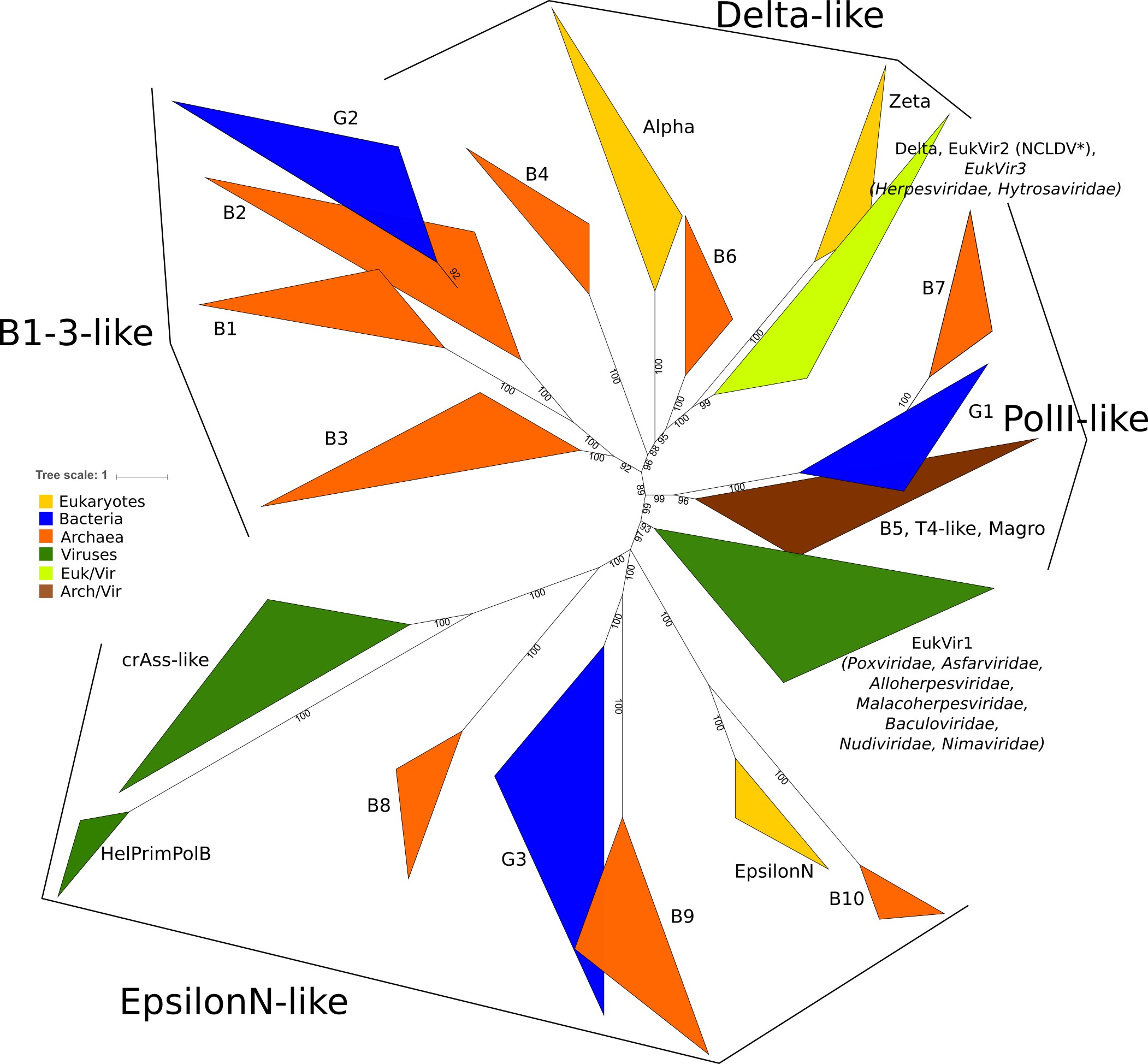
Phylogenetic tree of B-family DNA polymerases
(Kazlauskas et al. Nucleic Acids Res. 2020, 48: 10142–10156).
Surveying CARF and SAVED Proteins, Key Players in Antivirus Defence of Prokaryotes
Proteins possessing CARF and SAVED domains are key components of cyclic oligonucleotide-based antiphage signalling systems (CBASS) that, upon activation, induce cell dormancy or death. Most CARF proteins belong to a CBASS built into type III CRISPR–Cas systems. The CARF domain binds cyclic oligoA (cOA) synthesized by the Cas10 polymerase-cyclase and allosterically activates the effector, typically a promiscuous ribonuclease. Some CARF domains also function as ring nucleases that cleave cOA thereby terminating signal transduction. Due to the extreme sequence divergence and the diversity of domain architectures of CARF domain-containing proteins, CARF domains are often overlooked or misannotated in genome analyses. Therefore, we performed a comprehensive analysis of the CARF and SAVED domains encoded in bacterial and archaeal genomes. Based on this analysis, we proposed a classification of CARF and SAVED proteins, predicted several families of novel ring nucleases, and provided new insights into the organization of the cOA signalling pathway (Makarova et al. Nucleic Acids Res. 2020, 48: 8828–8847).
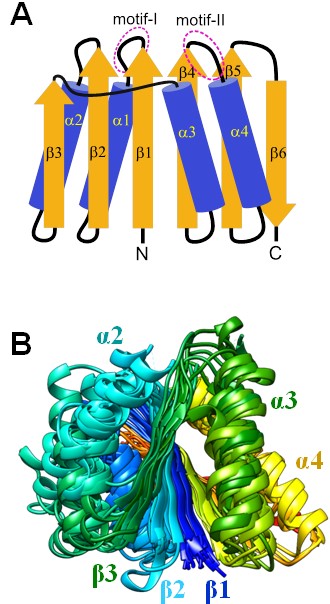
CARF domain structures (A) Topology of the CARF fold (B) Superposition of multiple CARF domain structures coloured by chain progression.