
Achievements of the AI program AlphaFold are mind-blowing, but it also has its limitations. “The program AlphaFold solved a problem that seemed insurmountable – it determines the structures of proteins and protein complexes by non-experimental methods. The program does it extremely accurately. […] Fortunately, AlphaFold is not yet all-powerful, because if it could do everything, we would be out of work”, jokingly says Dr. Giedrius Sasnauskas, a biochemist of Vilnius University Life Sciences Center (VU LSC).
Dr. Giedrius Sasnauskas is one of the few structural biologists in Lithuania. As he himself says, looking at the atomic worlds, he sees something that no one has seen before. Curiosity to examine nanometer-sized three-dimensional labyrinth-like objects led Dr. Sasnauskas and his colleagues to their success story – the determined structure of the evolutionary precursor of the CRISPR-Cas gene-editing tool.
|
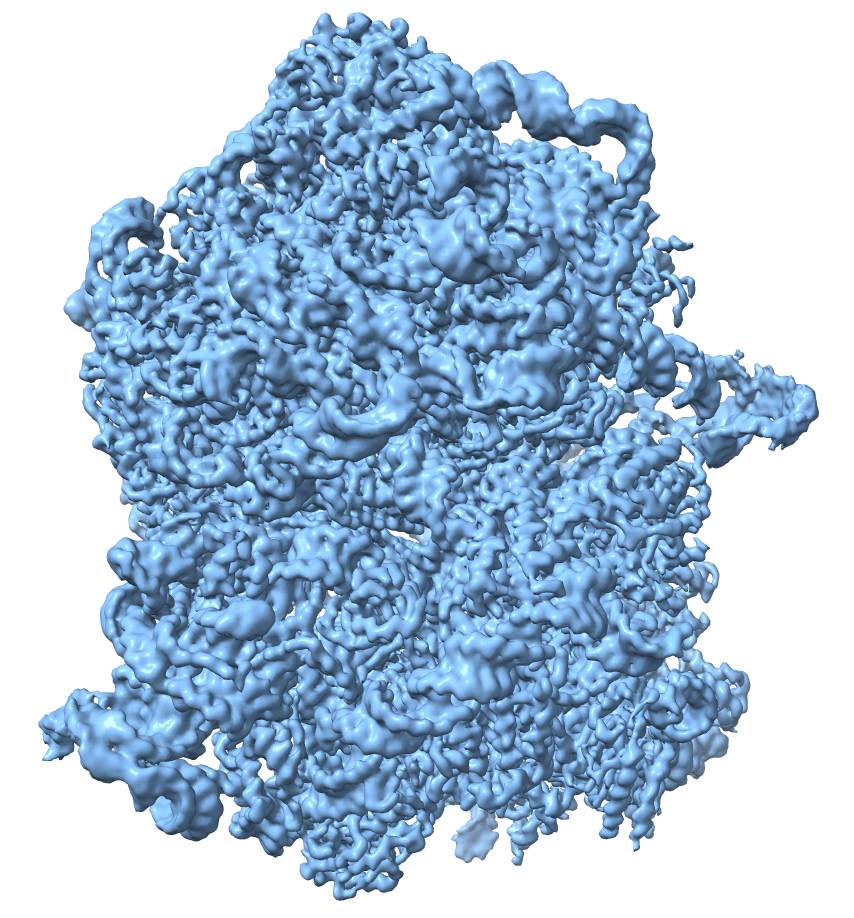
Cryo-EM map of a bacterial ribosome
|
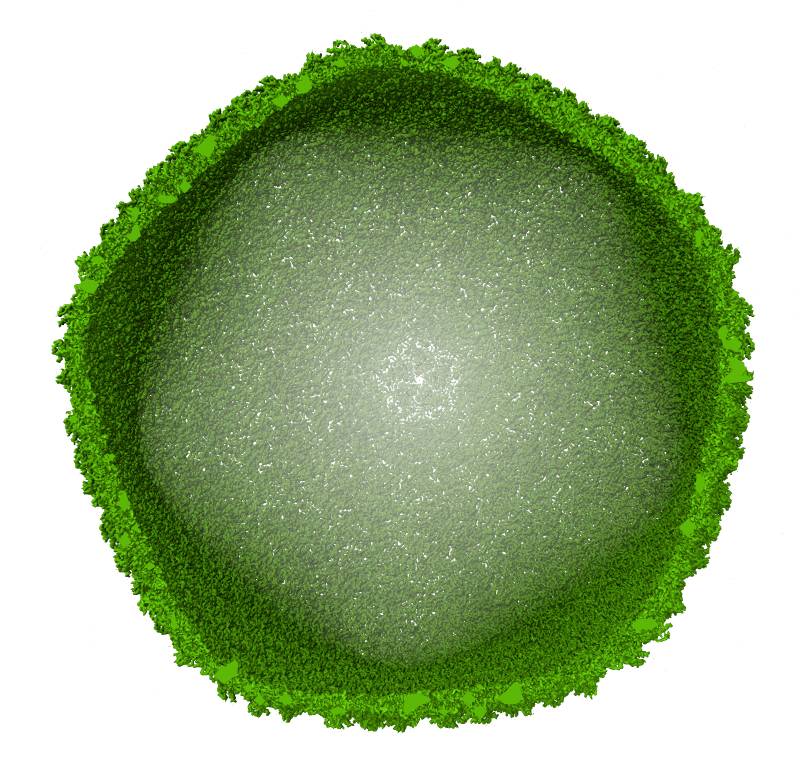
Cryo-EM map of a viral capsid
|
Q: What interests you about structural biology and how can the cryo-electron microscopy method you use help it?
A: Some people look at the stars, looking for something that no one has discovered yet. Someone may be traveling to mountains, or distant forests, looking for a plant or a beetle that no one has seen. And we, structural biologists, are traveling into the world measured in nanometers, that is, millionths of a millimeter, and are the first to see what the molecules making up living organisms look like. I see no less charm and even excitement in it.
In its broadest sense, structural biology is the science that deals with biological structures, from things visible to the naked eye or observable through an ordinary light microscope, such as individual cells or parts of cells to much smaller objects, such as individual molecules.
Modern structural biology mainly focuses on the structures of the molecules that make up living organisms, especially proteins, at the atomic level. That is, we determine how thousands of atoms that make up the protein of interest are arranged in space. A typical object of our research is from a few nanometers to a hundred nanometers in size, that is, from an individual protein to a small virus.
There are three experimental techniques used in structural biology: nuclear magnetic resonance, X-ray crystallography, and cryogenic electron microscopy, or cryo-EM in short. Despite being the latest addition, it is cryo-EM that is currently the most widely used technique for the determination of protein structures, owing to its efficiency and convenience. Thanks to the European Union funds, we have a cryogenic electron microscope at the Life Sciences Center of Vilnius University, and we have been using it intensively for three years now. With it, we can analyze large protein complexes and even viral particles.
Q: How do you obtain the structure of a protein using cryo-EM technique?
A: Sample preparation for cryogenic microscopy is quite an interesting process. The solution of the molecule of interest, for example, of the protein is frozen as very thin ice sheets with a thickness of several tens of nanometers. Protein molecules also freeze in that ice. Imagine clear ice several centimeters thick on the surface of the lake with leaves frozen in it. We have something similar, only reduced a million times.
We then take pictures of many of these thin ice sheets with an electron microscope, and from the resulting images, we extract the images of individual protein particles frozen in ice. And from many, tens or hundreds of thousands, sometimes even millions of images of such particles, we get a very detailed image of a protein molecule similar to a dense three-dimensional labyrinth. Later, we “wander” through that labyrinth and fill its “caves” with atoms that make up the protein. This gives us a model of the protein structure.
A fundamental advantage of cryo-EM compared to traditional electron microscopy is that the protein or another molecule frozen in ice remains in its native “wet” state. Freezing occurs very quickly, so it is possible to “catch” the protein as it is in solution. Samples in an ordinary electron microscope are dry; therefore, fine structural details of proteins are lost. And with cryo-EM, we see everything.
|
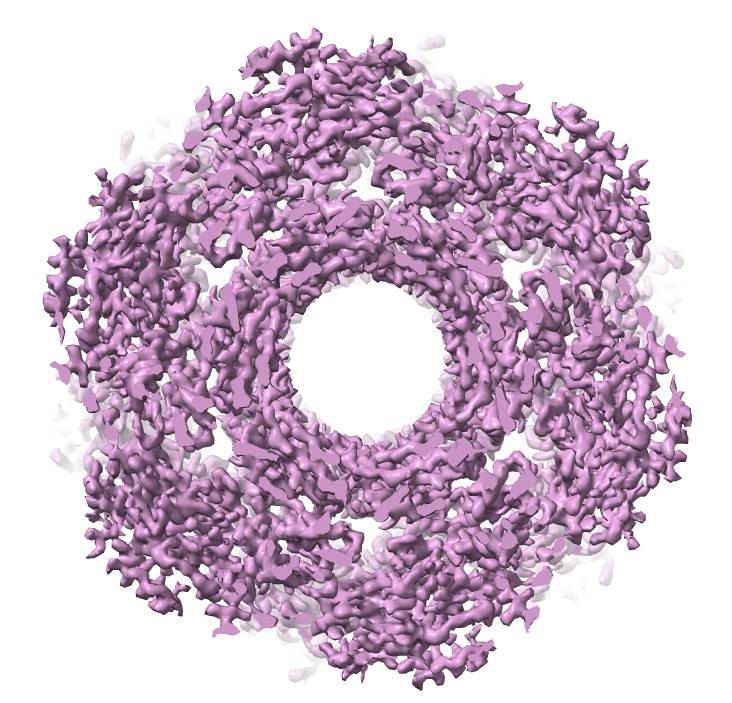
Cryo-EM map of a viral tail tube |
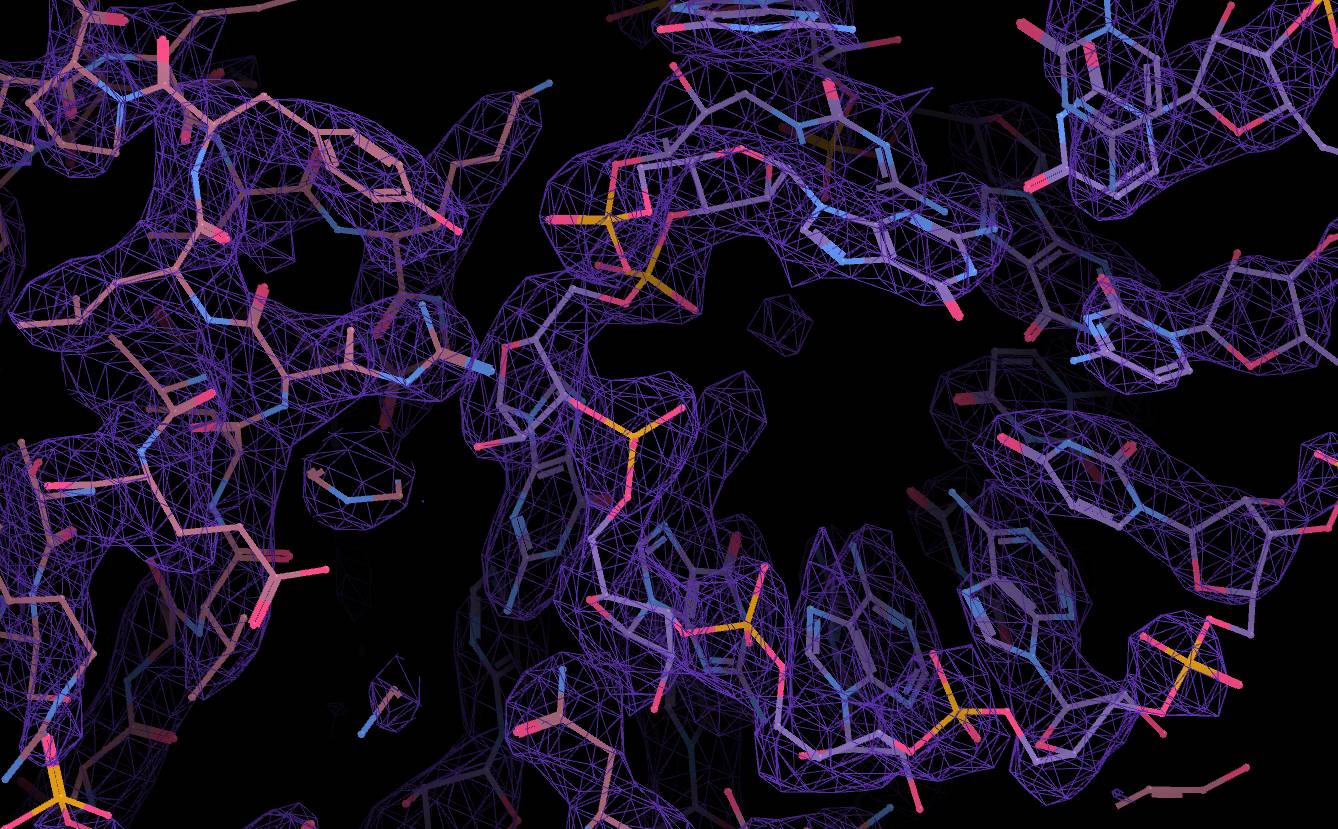
Structure model in a cryo-EM map |
Q: Artificial intelligence also helps to reveal protein structures. The AlphaFold app is getting quite a bit of attention. It is said to be able to build protein structures extremely quickly. What do you think of AlphaFold? Or are you using it?
A: For the past 15 years, we, structural biology researchers, have had a truly interesting life. During this time, several transformations took place.
First, one of the classic structural biology experimental methods, crystallography, has become much more accessible to many users. Second, the so-called “resolution revolution” in electron microscopy took place, when instead of low-resolution or low-detail images of molecules resembling one or several blobs stuck together, researchers could resolve finer details.
The final transformation was the AI-based algorithm AlphaFold 2. Published in the middle of 2021, it caused the revolution in less than a day. While many people encountered the possibilities of artificial intelligence only with the advent of ChatGPT, we, researchers working with protein structures, felt the power of artificial intelligence firsthand much earlier.
What did AlphaFold 2 do? It solved a problem that seemed unsolvable until then - it generates very accurate models of the structures of individual proteins or protein complexes and arranges thousands of atoms in space with angstrom accuracy.
Thus, AlphaFold does the same thing that we, structural biologists, conduct with cryo-EM and other experimental techniques. The first time we used AlphaFold we were blown away – the match of the AlphaFold model to our own experimentally determined protein structure was so good… (laughs). Then we got used to it. We now use the program as an auxiliary tool in our research.
Fortunately, the AlphaFold isn’t omnipotent just yet. Since the algorithm was trained on experimental protein structures, it successfully predicts namely the structures of many proteins. However, proteins often do not work alone but in combination with various non-protein components, for example, nucleic acids RNA and DNA, carbohydrates or smaller molecules. These components and their interactions with proteins are not modeled by AlphaFold, at least not yet. All experimental methods are still needed here, so we still have work to do. But I have no doubt that over time AlphaFold and similar algorithms will be able to do more and more.
Q: I would like to talk about the application of your findings. The structure of proteins is figured out, and where can it be used next?
A: It can be jokingly said that after determining the structure of a protein, the most important thing is to prepare beautiful pictures of it, which can be used to “decorate” a scientific publication. Protein structures are truly beautiful in their own way; they can even become art objects. On a more serious note, if we know how a particular protein is made, we can understand how it works. How it catalyzes some kind of chemical reaction or performs some other function.
To test our hypotheses about how a protein works, we can design and perform various experiments in the laboratory. And if we understand how the protein works, we can think about how to change its operation, to turn it in the direction we need.
Another very broad field of application of structural biology is drug design. In very general terms, if we have some kind of bacteria or virus attacking us, we can create a chemical compound that inhibits the action of some protein necessary for the “attacker” and thus acts as a drug. Structures of proteins with attached drug or drug fragment molecules provide a better understanding of how those drugs interact with target proteins and how those interactions can be further improved.
A good example is the recent SARS-CoV-2 virus pandemic. During it, researchers from various countries determined the structures of all viral proteins in a record time using structural biology methods. Thus, potential drug targets were identified as well. This knowledge also served in the development of vaccines.
Q: Before the interview, you mentioned that you have to share your latest discovery, and your success story. Tell us more, what did you discover?
A: As you probably already know, CRISPR-Cas9 and other gene editing scissors are our “pet project”. They made Lithuanian researchers famous in the world, but at the same time, they also helped popularize research in Lithuanian society. We, using cryo-EM, were able to determine the structure of a protein that is the evolutionary precursor of the CRISPR-Cas genome editing scissors.
This protein, called TnpB, is two to three times smaller and, in some ways, simpler than conventional CRISPR-Cas scissors. This spring, we published a paper about this structure in the Nature journal (doi: 10.1038/s41586-023-05826-x). It is a very important article for us, a true success story. Furthermore, it is the first published structure obtained with our cryo-EM microscope. That’s how we showed its capabilities.
Q: Why did you start looking for a precursor to CRISPR-Cas?
A: The so-called genome scissors, i.e. CRISPR-Cas9, CRISPR-Cas12, and similar proteins are found in bacteria, they help bacteria defend themselves against viruses called bacteriophages that attack them. The question arose as to how such complex enzymes evolved. It turned out that the precursors of CRISPR scissors originated in mobile genome elements called transposons.
Colleagues’ research has shown that a protein called TnpB encoded in the mobile genome element works very similarly to conventional CRISPR genome scissors but is much smaller. Therefore, we were interested in finding out how these tiny “primordial” genome scissors look like and how they work.
To this end, we have determined the structures of the TnpB protein using cryo-EM. We found that, although the TnpB protein is significantly smaller and simpler than CRISPR-Cas scissors, this is partially compensated by the large nucleic acid molecule bound to it. Therefore, TnpB performs the same function, that is, it recognizes a particular piece of DNA, binds it and cuts it.
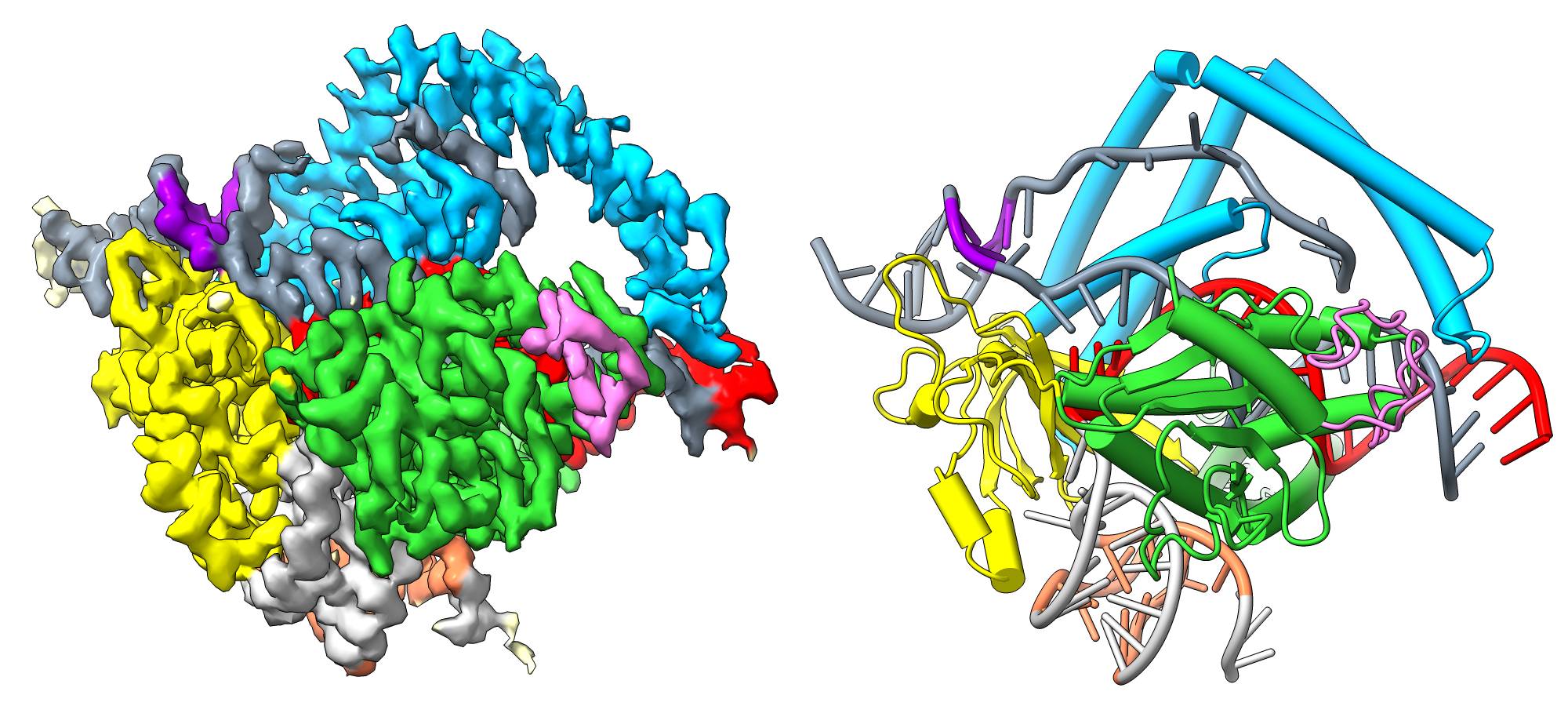
Q: How do you think the TnpB you examined could benefit humans? CRISPR-Cas9, just 12 years after its discovery, is already applied in treating sickle cell anemia, and research is continuing with other genetic diseases. Thus, clinical application appeared very quickly?
A: I cannot say exactly for what applications and methods these tiny genome scissors will be used. We already know several thousand genome scissors, the most popular being CRISPR-Cas9. But we have many different ones… Before researchers begin applying TnpB, it needs to be characterized more thoroughly.
For example, it is very convenient that it is a small protein, making it easier to transport it or its improved variants into cells. But the question is whether it is as accurate as other tools - does it indeed targets only the correct spot in the genome? If it cuts in the wrong spot, there is a risk of a new disease. So before application in practice, there is still a lot to be clarified.
Q: I am greatly interested to learn your opinion about communication with the public. Let’s say the concepts and terms discussed today, if most people just read them somewhere, these terms might not mean anything. But when you told it, you show how much is happening, how researchers are expanding the boundaries of our perception, and, as you say, they are seeing things that no one has seen before. It kind of makes sense. Do you yourself communicate with the public? Do you think it can help researchers in general?
A: I think the popularization of CRISPR-Cas9 in the Lithuanian public brought all sorts of benefits. Up to now, my contribution to public education has primarily comprised conducting tours at the VU LSC, mainly aimed at senior school students interested in natural sciences. During these tours, I introduce them to the field of structural biology including cryo-EM.
I have not yet been involved in the education of the wider society, though I do recognize the importance of sharing one’s work with the public. However, it’s not easy as communication skills need to be developed. After all, technical details and the routine of scientific work are really of little interest to anyone. You need to think about how to tell the story properly, and what comparisons to invent and present so that it is more accessible and engaging to everyone. In short, maybe this interview will be a great start.
Q: Thank you for your talk.
A: Thank you!
Interviewed by Goda Raibytė-Aleksa
Photo credits: Dr. Giedrius Sasnauskas' personal archive